

CAR-T in solid tumors (Part 2)
Unravel the scientific world of CAR-T cell therapy

Dear Readers,
This article is a continuation of a 4 part series on CAR-T. In Part 2 here, we will explore its opportunities and challenges in solid tumors.
A paradigm shift in oncology happened with the advent of synthetic immunity, i.e., adoptive engineered T cell therapy. In the past three decades, CARs have progressed from “bench to bedside” with FDA approvals of four CAR-T therapies for refractory or relapsed patients with haematological malignancies (details in Part 1: A deep dive into CAR-T). More CARs are currently undergoing clinical development for the treatment of other haematological entities, such as chronic lymphocytic leukaemia, or low grade lymphomas and myeloma (will be discussed in detail in Part 3: The CAR-T Innovators).
2.1 Why still no CAR-T for solid tumors ?
The efficacy of CAR-T cell therapy is unchallenged for a number of haematological malignancies, it is important to realise that, with over 100 types of cancer, haematological cancers comprise only a small fraction of diagnosed cancers and are responsible for only 6% of all reported deaths. Attempts to treat solid malignancies with CAR-T cells have shown minimal observable therapeutic benefit combined with elevated toxicities.
For effective CAR-T therapy, several factors are essential, such as recruitment, activation, expansion and persistence of bioengineered T cells at the tumour site.
Challenges in solid cancers
Solid tumors are difficult to tame and destruct, preclinical and clinical studies have identified multiple roadblocks which can be summarized in three major parts: Recognition, Trafficking and Survival of CAR-T cells in the solid tumor microenvironment.
The tumour microenvironment (TME) is acidic, hypoxic (deprived of oxygen), poorly vascularised (lack of conventional blood vessel hierarchy) and extracellular matrix (ECM)-rich which prevents T-cell infiltration inside tumors. If T cells reach tumor core they face difficulty in recognising antigens on tumors because of low immunogenicity. Furthermore, immunosuppressive environment, inhibitory surface proteins, cytokines or soluble products of disrupted cell metabolism within the tumour can also impair the activation and persistence of T cells. In summation, these extrinsic hurdles blunt the therapeutic efficacy of CAR-T cells in solid tumor setting.

Furthermore, the metabolic profile of the TME is highly unconducive to anti-tumour immunity. For CAR-T therapy to be effective, CAR-T cells need to rapidly proliferate, secrete cytokines and initiate killing of tumor cells—all of these tasks are metabolically demanding and CAR-T cells rely on rapid induction of aerobic glycolysis. Since TME is hypoxic and nutrient-deprived niche, cancer cells also primarily rely on aerobic glycolysis over the more energetically efficient mitochondrial oxidative phosphorylation to sustain biomass production. This dysregulated metabolic pathway is know as ‘Warburg effect’, named after the discoverer Otto Warburg.
CAR-T cells thus has to compete for glucose in the TME in an already nutrient-poor niche; but faces additional challenge as tumor cells are able to outcompete T cells. Glucose-limited CAR-T cells are unable to function as effectively in the TME, glucose deficiency leads to dampening of T cell receptor signalling and effector responses. Several strategies1 2 3 4 5 6 to rescue the glycolytic activity required for effector T cell function has been investigated and also showed promising results in pre-clinical trials.
In addition to the competition for limited fuel in TME, CAR-T cells are also challenged by immunosuppressive metabolites like indoleamine 2,3- dioxygenase (IDO)–tryptophan–kynurenine axis, adenosine, lactate, etc. These metabolites suppress the CAR-T effector function through multiple mechanisms. However, now with the advancement of basic research and technology, we better understand which metabolism directly affects T cell fitness and now can employ myriad of ways to reprogram T cells to calibrate their metabolic fluxes. The strategies can be accomplished via both genetic and pharmaceutical means. Some of these strategies7 8 9 has also shown promising results in both pre-clinical and clinical settings.

If interested to dig deeper: read Nature Metabolism Reviews: Metabolic barriers to cancer immunotherapy; Navigating metabolic pathways to enhance antitumour immunity and immunotherapy; Navigating CAR-T cells through the solid-tumour microenvironment
Opportunities in solid cancers
Solid tumors devise a hostile environment which protects them. Our goal is to develop multi-warhead missiles, against which cancer cells will surrender and find it difficult to develop resistance.
If your enemy is secure at all points, be prepared for him. If he is in superior strength, evade him. If your opponent is temperamental, seek to irritate him. Pretend to be weak, that he may grow arrogant. If he is taking his ease, give him no rest. If his forces are united, separate them. If sovereign and subject are in accord, put division between them. Attack him where he is unprepared, appear where you are not expected-Sun Tzu, The Art of War (This strategy can be well employed to win the battle against cancer)
Results from first in human clinical studies with CAR T cells (autologous CAR T cells targeting α-folate receptor) for ovarian tumors in 2016 showed safety but had rather disappointing anti-tumor responses and low persistence of infused T cells. Another study with CAR T cells targeting CD171 in neuroblastoma (cancer that develops from immature nerve cells) patients demonstrated T cell infusion but was not maintained. In 1999, Waldemar Debinski discovered that interleukin 13 (IL-13) was present on glioblastomas, but not on normal brain tissue, making IL-13 a good target to specifically target brain tumor cells. Currently six trials with IL13Ralpha2-CAR-T cells in glioblastoma patients are ongoing.
First-gen CAR-T cells directed against several antigens expressed by solid tumors: carbonic anhydrase IX (CAIX), CD171, folate receptor alpha (FR-α), GD2, human epidermal growth factor receptor 2 (HER2), mesothelin, EGFRvIII or vascular endothelial growth factor receptor 2 (VEGF-R2) did not significantly benefit patients. They show limited activity and frequent toxicity. Limited T cell persistence in these first trials is most likely due to several factors, including insufficient T cell activation due to lack appropriate co-stimulation in first gen CARs.
Different co-stimulatory molecules have been added to enhance CAR-T cell activation, these CARs are referred to as second generation (one co-stimulatory endodomain) or third generation (two co-stimulatory endodomains). Studies are done to evaluate the efficacy of third-generation GD2-CAR T cells with a CD28.OX40.ζ endodomain in patients with neuroblastoma. While infusion of CAR-T cells was safe, their anti-tumor activity was limited.
2.2 Navigating CAR-T cells through Solid TME
Preclinical and clinical studies have highlighted that no universal CAR construct for all malignancies exists, but rather each CAR must be optimized for the targeted antigen and each tumor setting.

Some of the optimisation techniques which can be employed to overcome obstacles for the treatment against solid tumors:
Optimizing CAR design by using 4-1BB co-stimulation in cis or trans
Expressing signal converters or dominant negative receptors
Improving homing to tumors by expression of distinct chemokine receptors
Promoting expansion and persistence of infused T cells using cytokines or cytokine receptors
Overcoming antigen heterogeneity or antigen loss by expression of CARs targeting two tumor antigens
Selecting T cell subsets for genetic modification
Target antigen choice
Antigen choice is a major determinant of safety and efficacy for CAR-T cell therapy. In principle, the ideal tumour antigen should be highly and uniformly expressed on tumour cells but absent on healthy tissue. However, the identification of suitable tumour antigens has been a longstanding challenge, even CAR-T therapies targeting the pan-B cell marker CD19 is successful in treating cancer but invariably results in damage of normal B cells. The strategy is to keep the delicate balance between eliciting potent anti-tumour activities and preventing severe off-target toxic effects.
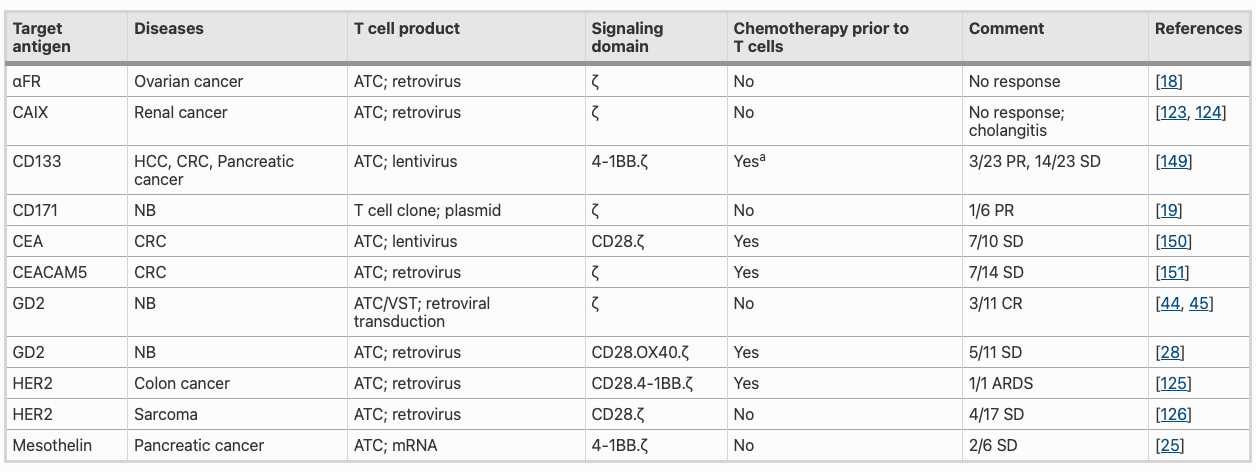
Here it’s very important to understand that even after careful choice of tumor antigen (like MAGE, which is not expressed on healthy cells) certain unanticipated off-target toxic effects were observed in clinical studies. MAGE-A3-targeted TCR T cells therapy resulted in severe neurotoxicity in two patients resulting in fatality. This was attributed to cross-reactivity of the MAGE-A3 TCR with unanticipated MAGE-A12 expression in the brain. Mesothelin has emerged as a promising tumor-associated antigen for solid tumours given its over-expression in various solid tumours and its limited expression in healthy mesothelial cells. Despite a favourable safety profile, mesothelin-targeted CAR-T cells have shown limited efficacy in clinical trials as a monotherapy. Recent clinical trials have shown that combining CAR-T therapy with monoclonal antibodies targeting checkpoint inhibitors or changing the delivery profile can increase the efficacy of therapy. There is a lot of room for improvement that we will be discussing throughout this article.
2.3 Toward a perfect CAR T
Given the risk of targeting antigens that are associated with, but not exclusive to, tumour cells, several engineering strategies have been developed to improve the tumour-targeting specificity of CAR-T cells. Some of these strategies we have discussed earlier. Let’s go through them in detail here:
Strategy is to fine-tune the affinity of CARs to their cognate antigens:
AND CAR-Signal complementation
AND- gated antigen recognition where each of two receptors must engage its own cognate antigen for a full T cell signalling response to occur.
SynNotch-induced CAR
In synNotch-induced CAR expression, a constitutively expressed synthetic Notch (synNotch) receptor triggers CAR expression upon cognate antigen binding. The target antigen of the CAR must also be present to activate the engineered T cells.
AND or AND-NOT SUPRA CAR
AND-NOT gated computation is achieved can be achieved through the split, universal and programmable (SUPRA) CAR platform, which consists of zipFvs (scFv sequences fused to leucine zippers) and T cells expressing zipCARs (CARs with extracellular domains consisting of a leucine zipper).
AND-gate CARs activate under a prevalent characteristic of the TME, like hypoxia
Single-chain bispecific CAR
encodes two ligand-binding domains in a single receptor, or by co-expressing multiple receptor chains in a single T cell, CAR-T cells can be programmed to target multiple tumour antigens.
Multi CAR
Layered approach to multi-antigen targeting to reduce the risk of off-tumour toxicity. CARs specific to target neoantigens (novel epitopes generated through patient-specific tumor mutations) and intracellular targets. Now with whole-exome sequencing of tumour biopsies, neoantigens can be computationally predicted. However, neoantigen based therapies success is limited to cancers with high mutational burden.
For further reading, check the review by Yamamoto and colleagues and work by Peng and colleagues on neoantigens.
2.4 Grafting CARs on T Cell Subsets
In solid tumor settings, preferentially selecting a defined CD8+ and CD4+ subset prior to infusion could also lead to enhanced antitumor efficacy. Approaches like generating CAR-T cells from Th17 and Tc17 cells will be more potent compared to conventional approach. The reason being, these cells have better survival capabilities and are known to have longer persistence in the TME.
Additionally, CD26 high CAR-T cells have also been investigated because they have superior function in solid tumour models as a result of increased cytokine production memory, stem-like properties and trafficking. γδ T cells are another potentially advantageous subset. The vast majority (95–99%) of typical CAR-T cell infusion products are αβ T cells. Both subsets show equivalent levels of cytotoxicity; however, γδ CAR-T cells are less prone to exhaustion. In addition, γδ CAR-T cells express co-stimulation and antigen-presentation molecules and have been shown to cross-present tumour antigens to other T cells in the TME.

2.5 Allogenic (Off-the-shelf) CAR T cell
In comparison to traditional autologous, universal off-the-shelf CAR-T will improve standardization of the product, patient waiting times, logistics around coordinating manufacturing with patient urgency and cost. The main risks associated with allogenic CARs are GvHD (Graft versus Host disease) which pose safety and efficacy issue.
The first clinical trial with allogenic CAR-T were published in 2017, when two infants with B-ALL were treated with TALEN-edited TCRα-negative, CD52-negative 4-1BB-based CD19-targeted CAR-T cells. Both patients showed complete remission with minimal GvHD. Several ongoing trials with allogenic CAR for adult ALL are ongoing. Other allogeneic CAR-T cell products involve multiplexed-genome editing via CRISPR (Clustered Regulatory Interspaced Short Palindromic Repeats)–Cas9 technology. Due to its scalability, affordability and ease of use this gene-editing method has become the gold standard. Another gene editing approach, such as TALENs (Transcription Activator-Like Effector Nucleases) and ZENs (zinc finger nuclease) have seen significant advances and are now being employed to design advanced CARs. Advancements are being made for automated T cell engineering process to produce off-the-shelf or universal CAR-T cells on an extended CliniMACS Prodigy platform containing an in-line electroporation unit.
2.6 CAR-T cell safety and control
Safety considerations while designing CAR-T for solid tumours are critical. In light of the toxic effects commonly associated with CAR-T cell therapy, mitigation strategies and engineered safety controls are active areas of research. Some of the strategies are discussed below:
Implement suicide genes into CAR-T cells that lead to cell death upon small molecule-mediated induction of the suicide gene,
The STOP-CAR system which incorporated domains that enables drug-mediated pausing of CAR-T cell activity,
The GoCAR-T system enables drug-mediated activation of CAR-T cell activity.

CAR-T cell research advanced rapidly for blood cancers into the clinic and now its back to bench to find ways to combat solid tumors. Innovations in CAR design, transduction methodologies and selection of the best cell types are bound to lead to improved responses and transform the treatment of patients with many different cancer types. It can be rightly said that road towards the broader and more successful application of CAR-T cell therapy is currently being paved.
In Part 3 of this series, we will examine the translational world of CAR-T and its worldwide progress. Stay tuned.
Please sign up and spread the word. Help me help you to better grasp this space.
Qiu, J. et al. Acetate promotes T cell effector function during glucose restriction. Cell Rep. 27, 2063–2074.e5 (2019).
Renner, K. et al. Restricting glycolysis preserves T cell effector functions and augments checkpoint therapy. Cell Rep. 29, 135–150.e9 (2019).
Vardhana, S. A. et al. Impaired mitochondrial oxidative phosphorylation limits the self- renewal of T cells exposed to persistent antigen. Nat. Immunol. 21, 1022–1033 (2020).
Dumauthioz, N. et al. Enforced PGC-1α expression promotes CD8 T cell fitness, memory formation and antitumor immunity. Cell. Mol. Immunol. https:// doi.org/10.1038/s41423-020-0365-3 (2020).
Ligtenberg, M. A. et al. Coexpressed catalase protects chimeric antigen receptor–redirected t cells as well as bystander cells from oxidative stress–induced loss of antitumor activity. J. Immunol. 196, 759–766 (2016).
Gemta, L. F. et al. Impaired enolase 1 glycolytic activity restrains effector functions of tumor- infiltrating CD8+ T cells. Sci. Immunol. 4, eaap9520 (2019).
Triplett, T. A. et al. Reversal of indoleamine 2,3-dioxygenase–mediated cancer immune suppression by systemic kynurenine depletion with a therapeutic enzyme. Nat. Biotechnol. 36, 758–764 (2018).
Beavis, P. A. et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J. Clin. Invest. 127, 929–941 (2017).
Angelin, A. et al. Foxp3 reprograms T cell metabolism to function in low- glucose, high- lactate environments. Cell Metab. 25, 1282–1293.e7 (2017).